Highlighting some releases in July 2023

We wanted to highlight three updates that we made in July 2023.
First, Exon now will rewrite queries to scan multiple files in parallel. This enables considerable speedups for queries over large datasets -- both for local files and object stores (e.g. S3, GCS, etc.).
Second, we're releasing an initial version of an MzML reader in our suite of tools. This is our first step towards supporting proteomics and metabolomics data in exon. This format is subject to change as we get feedback from users, so please let us know what you think!
Third, we're adding support for pandas dataframes in BioBear. This is a common request, so we're excited to be able to support it.
Parallel Scan
Not only is going fast fun, but it's also important for enabling the fast iteration cycles necessary for innovation. To that end, Exon and related tools will now automatically parallelize scans over multiple files.
For example, if you have a directory with FASTA files, you can now do the following:
import biobear as bb
from pathlib import Path
data = Path("./data/") # folder with fasta files
# note: FastaReader is now deprecated, use session.read_fasta_file(data)
df = FastaReader(data).to_polars() # need to install polars
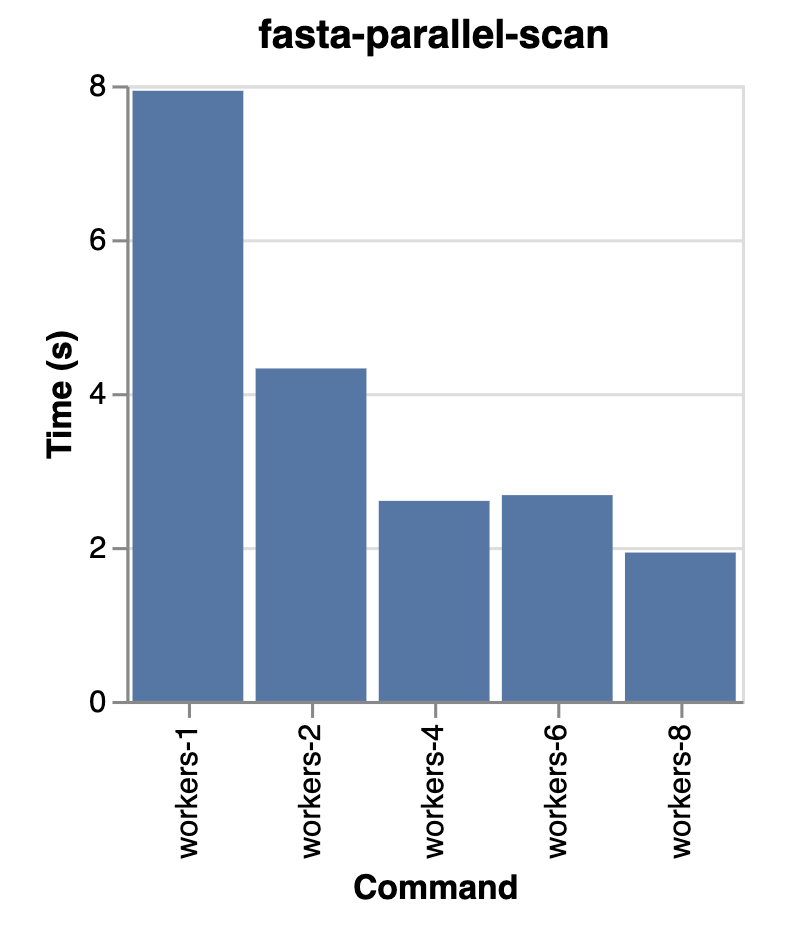
And Exon will distribute reading the files across available cores. This is a huge win for data lakes, where you can now read multiple files in parallel without having to manually shard your data. E.g. here we see a 4x speed-up when reading 8 files.
MzML Reader
MzML is a very common format in metabolomics and proteomics. At its core are measurements of the spectrum from the mass spec, but there's also a Controlled Vocabulary that describes various metadata about the experiment.
Similar to other readers, you instantiate a reader and then depending on what you want to do with it, you can convert it to arrow or polars with BioBear, or a data.frame with R.
import biobear as bb
# object stores and local files are supported
df = bb.MzMLReader("mzml-data/GNPS00002_A3_p.mzML").to_polars()
df.head()
# shape: (5, 6)
# ┌───────────────────────────────────┬───────────────────────────────────┬───────────────────────────────────┬────────────┬───────────────────────────────────┬────────────────┐
# │ id ┆ mz ┆ intensity ┆ wavelength ┆ cv_params ┆ precursor_list │
# │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │
# │ str ┆ struct[1] ┆ struct[1] ┆ f32 ┆ object ┆ object │
# ╞═══════════════════════════════════╪═══════════════════════════════════╪═══════════════════════════════════╪════════════╪═══════════════════════════════════╪════════════════╡
# │ controllerType=0 controllerNumbe… ┆ {[80.948143, 80.993134, … 957.16… ┆ {[5416.88916, 7712.663574, … 480… ┆ null ┆ [('MS:1000579', {'accession': 'M… ┆ None │
# │ controllerType=0 controllerNumbe… ┆ {[80.284782, 81.017242, … 1176.3… ┆ {[4701.711426, 5527.709961, … 95… ┆ null ┆ [('MS:1000579', {'accession': 'M… ┆ None │
# │ controllerType=0 controllerNumbe… ┆ {[80.948288, 81.017365, … 1193.9… ┆ {[11152.501953, 9900.050781, … 8… ┆ null ┆ [('MS:1000579', {'accession': 'M… ┆ None │
# │ controllerType=0 controllerNumbe… ┆ {[80.948143, 81.017372, … 1074.1… ┆ {[6677.85791, 11401.181641, … 12… ┆ null ┆ [('MS:1000579', {'accession': 'M… ┆ None │
# │ controllerType=0 controllerNumbe… ┆ {[80.948174, 81.017387, … 968.84… ┆ {[6586.833984, 13542.833008, … 6… ┆ null ┆ [('MS:1000579', {'accession': 'M… ┆ None │
# └───────────────────────────────────┴───────────────────────────────────┴───────────────────────────────────┴────────────┴───────────────────────────────────┴────────────────┘
For R users, see the read_mzml_file function. And for exon-duckdb users see
Please see the docs for more information about MzML files, and again, please let us know if you have any feedback!
A Sneak Peak for Querying
While it's not quite ready yet for BioBear and ExonR, in Exon, we've added a set of MzML specific functions for querying. For example, you can filter spectra based on the presence of a specific peaks.
For example, this will filter a set of spectra to only those that have a peak at 100.0 m/z that's within 0.1 m/z of the peak.
SELECT id
FROM mzml -- same table as above
WHERE contains_peak(mz.mz, 100.0, 0.1) = true
This sort of declarative informatics is part of the Exon vision, which we'll be talking more about in the future.
A Note on Performance
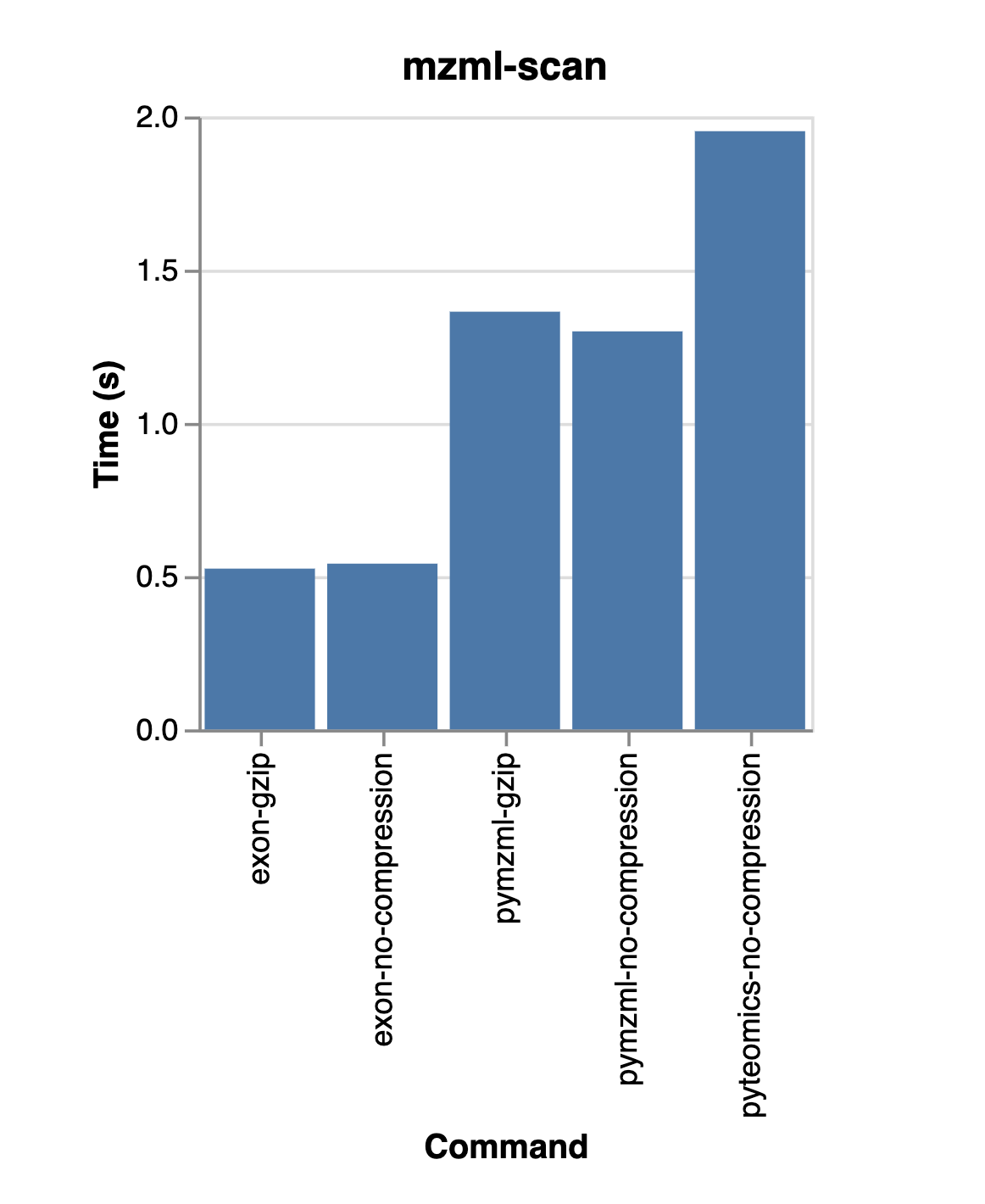
Similar to BioBear comparing favorably to other scientific data libraries, we see modest out-performance for MzML file reading.
The following compares reading using a few difference libraries in Python on an MzML file with about 8k spectra.
Pandas Support in BioBear
There's not much to say besides that places where you could use to_polars you can now also use to_pandas. E.g.
import biobear as bb
from pathlib import Path
data = Path("./data/uniref50.fasta")
# note: FastaReader is now deprecated, use session.read_fasta_file(data)
df = FastaReader(data).to_pandas() # need to install pandas
df.head()
# id description sequence
# 0 UniRef50_A0A5A9P0L4 peptidylprolyl isomerase n=1 Tax=Triplophysa t... MEEITQIKKRLSQTVRLEGKEDLLSKKDSITNLKTEEHVSVKKMVI...
# 1 UniRef50_A0A410P257 Glycogen synthase n=2 Tax=Candidatus Velamenic... MKAIAWLIVLTFLPEQVAWAVDYNLRGALHGAVAPLVSAATVATDG...
# 2 UniRef50_A0A8J3NBY6 Gln_amidase domain-containing protein n=2 Tax=... MEILGRNLPRILGNLVKTIKTAPVRVVARRGARTLTQKEFGKYLGS...
# 3 UniRef50_Q8WZ42 Titin n=3053 Tax=cellular organisms TaxID=1315... MTTQAPTFTQPLQSVVVLEGSTATFEAHISGFPVPEVSWFRDGQVI...
# 4 UniRef50_A0A401TRQ8 Ig-like domain-containing protein (Fragment) n... PPSFIHKPDPQEVLPGSNVKFTSVVTGTAPLKVSWFKGTTELVAGR...
Conclusion
We hope you enjoy the new features and please let us know if you have any feedback! We'll be back next month with more updates.